ChIP-Atlas Update: 10th Anniversary of a Data-Mining Platform for Epigenome Exploration
- Others
- Funding
- Database Integration Coordination Program
On Apr 29, 2026, Professor Shinya Oki of the Institute of Resource Development and Analysis at Kumamoto University, and his colleagues, published a paper to mark the 10th anniversary of ChIP-Atlas and a significant update to a special web server issue of the scientific journal Nucleic Acids Research. In addition, on May 8, Kumamoto University, Chiba University, RIKEN, and the National Institute of Genetics issued a joint press release .
.
ChIP-Atlas is an integrated epigenomics database containing genome-protein interaction data (ChIP-seq), open chromatin data (DNase-seq and ATAC-seq), and DNA methylation data (Bisulfite-seq) for human and five model organisms (mice, rats, fruit flies, nematodes and budding yeasts). Since its release in 2015, ChIP-Atlas has been used across many research fields--such as genetics, studies of disease mechanisms, drug discovery, and developmental biology--and has been cited in over 1,500 domestic and international publications.
This update introduces a new feature that visualizes the reliability of individual experimental datasets. It shows how the read and peak counts of each dataset compare to those of similar experiments. It helps users to assess whether an experiment is supported by sufficient sequence reads and peaks to be considered reliable. Furthermore, users can evaluate dataset similarity and determine whether results are typical or exhibit distinctive patterns by comparing a dataset with others conducted under similar conditions.
ChIP-Atlas provides a transcription factor enrichment analysis which integrates hundreds of thousands of experimental datasets and performs cross-sectional data mining in order to predict gene expression regulatory factors. Previously, the enrichment analysis examined transcription factors that commonly bind to lists of differentially expressed genes (DEGs) provided by users, thereby identifying upstream factors that collectively regulate those genes. In this update, a new PAGE-based enrichment module has been added, which accepts RNA-seq gene expression data as input.
This module treats changes in the expression of all genes as continuous values and evaluates the overall directional change of each transcription factor's target gene set. This enables the detection of regulatory factors affecting genes with modest expression changes, which threshold-dependent DEG approaches might miss.
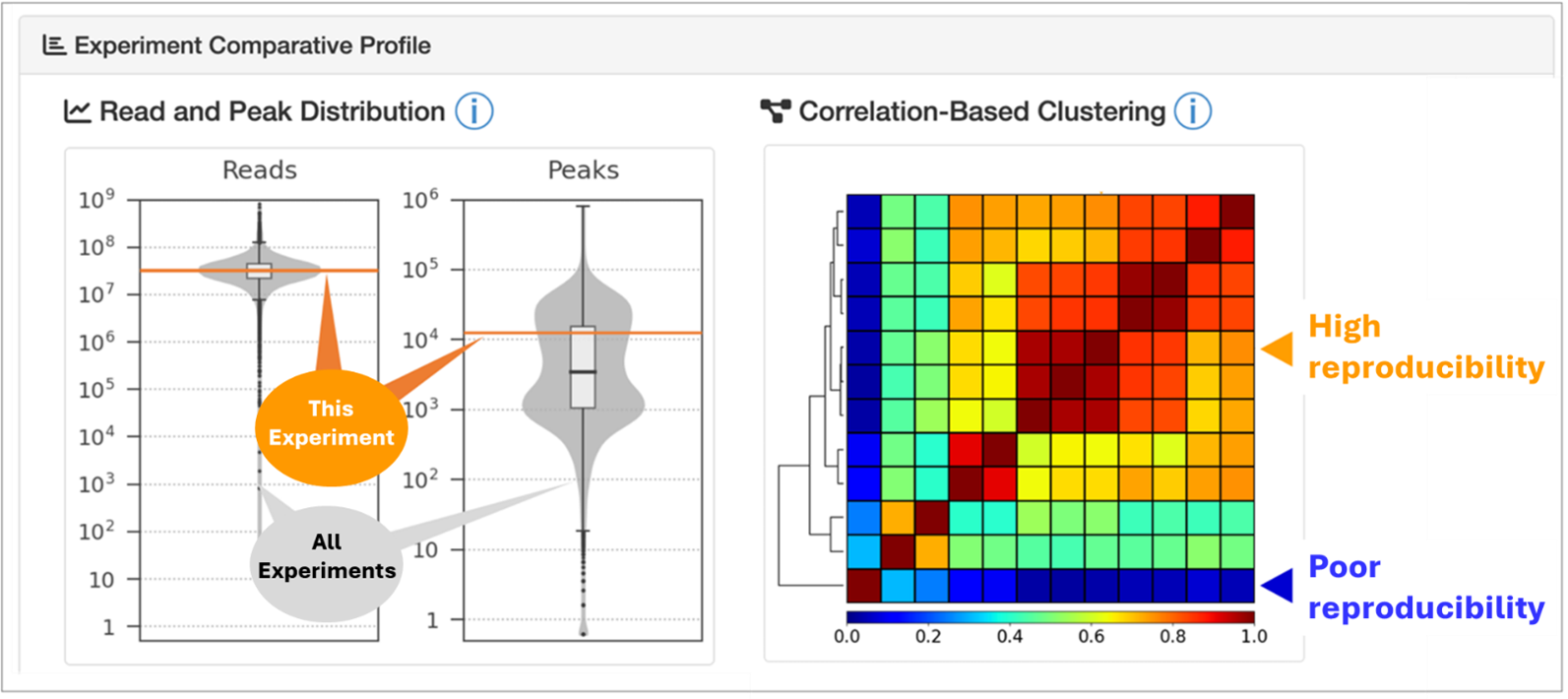
Figure 1. Experimental Data Quality Control Framework
Quality-control panel for SRX10829255 (https://chip-atlas.org/view?id=SRX10829255). (Left) The read- and peak-count distribution plot shows the position of the selected experiment (orange horizontal line) relative to comparable datasets. (Right) The correlation heatmap displays pairwise Pearson correlations between experiments, with colors from blue (low) to red (high). Hierarchical clustering groups experiments by similarity of genome-wide signal profiles. Arrows indicate the selected experiment(s); color denotes the median correlation with other experiments in the same biological context.
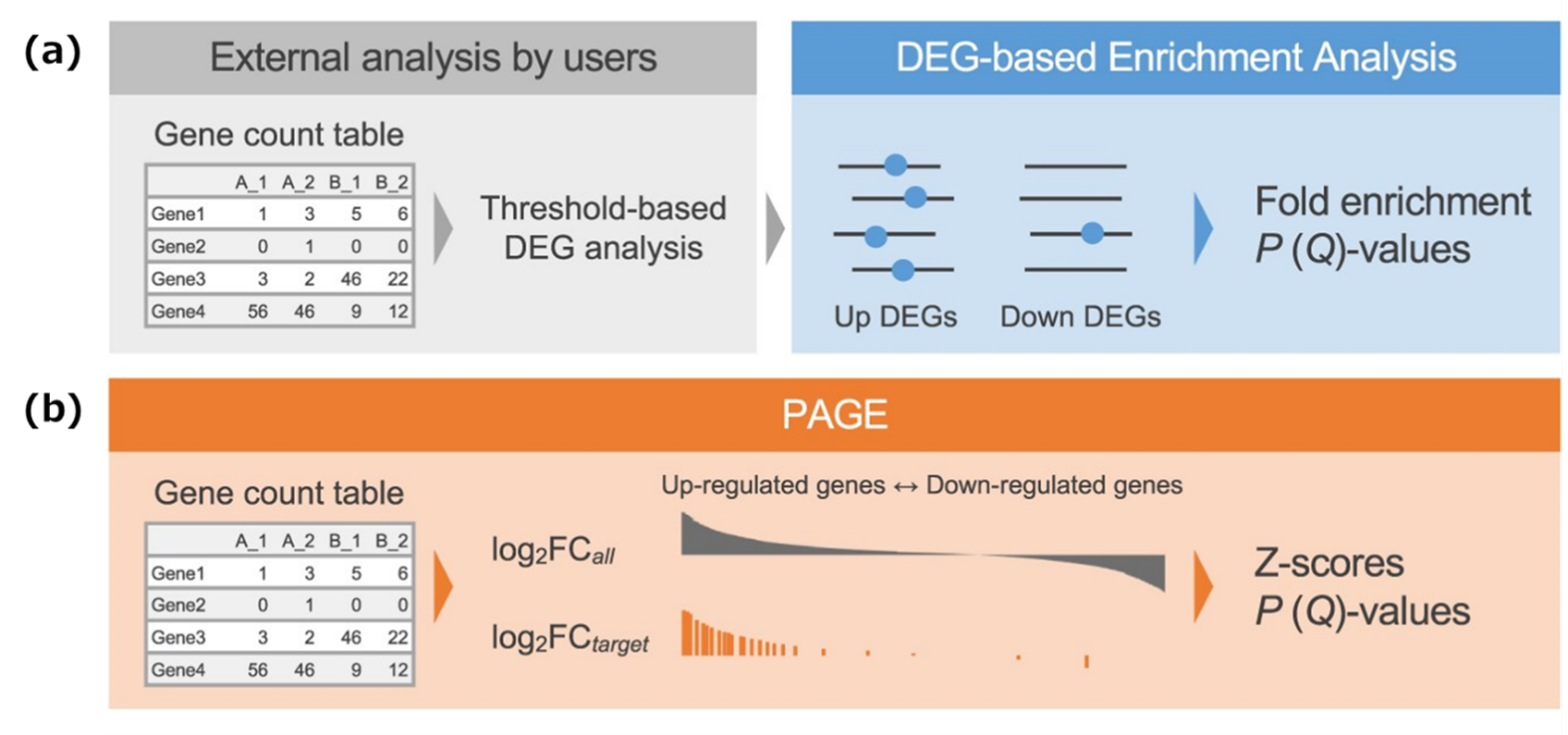
Figure 2. Schematic diagrams of DEG-based and PAGE-based enrichment analysis
(a) Schematic diagram of DEG (differentially expressed gene)-based enrichment analysis. It comprehensively examines transcription factors that commonly bind to a list of genes with different expression levels (DEGs), and identifies and presents upstream factors that collectively regulate them. (b) Schematic diagram of PAGE (parametric analysis of gene set enrichment)-based enrichment analysis. PAGE uses expression values for all genes and compares the mean log₂ fold change of genes in each ChIP-seq target set with the genome-wide mean to compute a Z-score quantifying gene-set-level transcriptional enrichment. This allows for the analysis of regulatory factors that reflect gene groups that undergo coordinated transcriptional changes even with small fluctuations, which may be missed in threshold-dependent DEG analysis.
ChIP Atlas is being developed as part of the JST Program for Database Integration Coordination Program (DICP), "Construction of an integrative data platform for transcriptional regulations" (Principal Investigator: KASUKAWA Takeya, Team Leader, Center for Integrative Medical Sciences, Riken).
For more information, see the paper and the press release.
<ChIP-Atlas data> (as of May 1, 2026)
| ChIP-seq (genome-protein interactions) | 262,634 entries |
| ATAC-seq (open chromatin information) | 97,673 entries |
| DNase-seq (open chromatin information) | 6,495 entries |
| Bisulfite-seq (methylome information) | 65,516 entries |
| Total | 432,318 entries |
Related Links
- Press release "Decades of Epigenetic Evolution: Unraveling the "Command Center" of Gene Expression Regulation" | Kumamoto University
- "ChIP-Atlas 2025 Update: 10-year anniversary of a data-mining platform for exploring epigenomic landscape" | Nucleic Acids Research
- ChIP-Atlas
- INTRARED
- Construction of an integrative data platform for transcriptional regulations | NBDC
Project summaries and reports are posted.

